
 |
|
||||
BiographyChristoph Wilhelmer was born 1993 in Lienz, Austria. He received his Bachelor degree in Technical Physics from TU Wien after doing an exchange semester at the University of Bath, UK in 2017. He received his Diplom-Ingenieur degree in Technical Physics from TU Wien in 2020 where he carried out his Master Thesis in the Quantum Materials group of the Institute of Solid State Physics. In March 2020 he joined the Institute for Microelectronics where he started his PhD, focusing on defect creation mechanisms in oxides used for semiconductor applications. |
|||||
Charge Transition Barriers of Hydroxyl-E' Centers in the Harmonic Approximation
It is well established that oxide defects are responsible for reliability issues in modern electronic devices by trapping charges from the substrate during operation. While for most experiments only either the electron or the hole capture process is relevant, some scenarios exist, like switching a transistor between accumulation and inversion mode, where both processes must be considered simultaneously, and even direct transitions between the negative and the positive charge states might be possible. In the amorphous oxide of MOSFET devices, defect parameters such as formation energies, trap levels, and relaxation energies are distributed over a wide range due to the stochastic ordering of atoms in the structure. The hydroxyl-E’ center (H-E’), which is formed during H annealing of oxidized Si surfaces by breaking strained Si-O bonds, is a defect candidate of particular interest due to its high concentration in the oxide and the close vicinity of its trap levels to the band edges of Si and SiC substrates. H-E’ configurations are shown in the neutral, positive, and negative charge state in Fig. 1, with the electron localized at the defect site.
We use Density Functional Theory (DFT), which has been proven to be a trustworthy tool over the last decades, to calculate structural and electronic properties of solids, and to analyze the H-E’ by calculating a distribution of total energies and relaxation energies in a-SiO2 in different charge states. The potential energy surface (PES) of a defect structure can be approximated near the energy minimum of the defect by fitting a parabola to the calculated minimum energy and relaxation energies as shown for an H-E’ in the three charge states and for different Fermi levels in Fig. 2. Subsequently, the energy barriers for transitions involving charge transfer, which correspond to the crossing points of PES in different charge states, can be extracted for distinct Fermi levels.
The barrier distributions for different charge exchange processes are shown in Fig. 3 as a function of the Fermi level. The charge transfer processes involving two charge carriers (-/+ and +/-) have significantly higher transition barriers compared to simple hole or electron capture, which makes this process energetically less favorable, meaning that a positively charged defect preferably becomes neutral before trapping another electron.
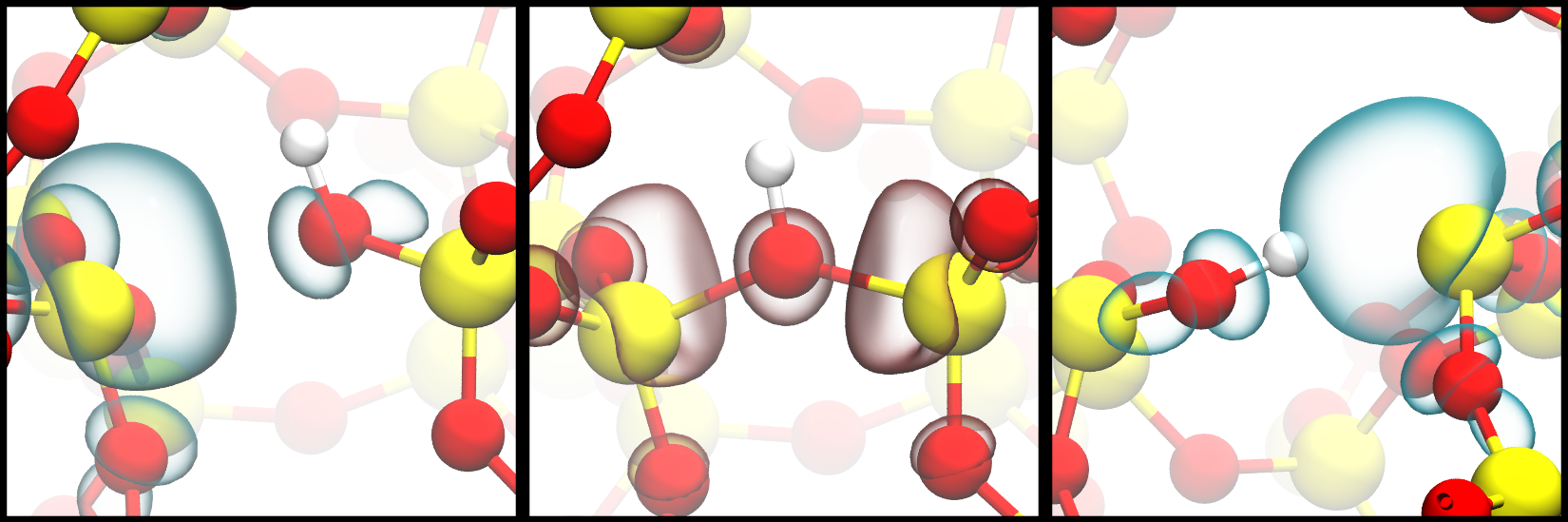
Fig. 1: Configurations of Hydroxyl-E' defects in the neutral (left), positive (middle), and negative (right) charge state, with the electron localized at the defect site.

Fig. 2: Potential energy surfaces in the harmonic approximation of a hydroxyl-E' defect in different charge states as a function of the normalized configuration coordinate for three distinct Fermi levels. Minimum energies and relaxation energies for all charge states are calculated with DFT. A parabola was fitted to these defect parameters to extract the transition barriers from the crossing points (transition states TST) of the PESs.
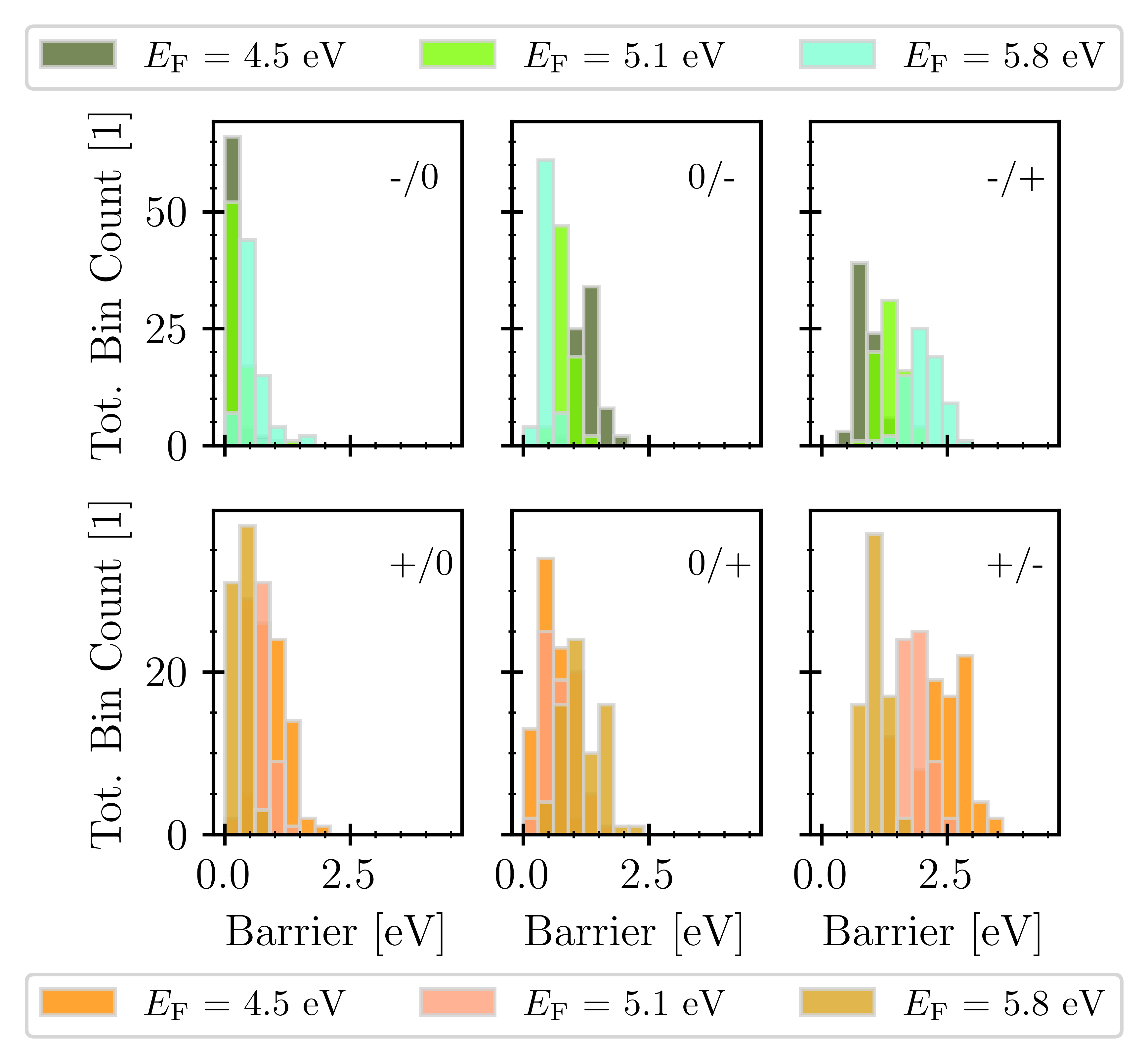
Fig. 3: Distributions of transition barriers of hydroxyl-E' defects for different charge exchange processes shown for three distinct Fermi levels.