
 |
|
||||
BiographyMohammad Dehghani is from Isfahan, Iran, where he has finished his bachelor's and Master's degrees in Solid-state Physics. In late 2013, he moved to Leoben, Austria and started working as a research assistant at Materials Center Leoben (MCL) Forschung GmbH. After receiving his PhD in computational solid-state physics from the University of Graz in late 2017, he continued working at MCL as a Postdoc for two more years. His scientific and research background focuses on the Density Functional Theory (DFT) simulations of inter-metallic alloys. He has years of experience working on projects focused on computational code development and application on various alloys. He has joined the Institute for Microelectronics in January 2023 and is working on ab initio simulation of quaternary semiconductor alloys. |
|||||
Ab initio Calculation of Phonon-limited Carrier Mobility in Semiconductor Alloys
Understanding carrier mobility is paramount in today’s semiconductor industry due to its direct impact on the efficiency of electronic devices. This property profoundly influences the performance of semiconductor devices such as transistors and optical devices, e.g., diodes and photodetectors. In an era of advancing semiconductor technologies with more complex functional material outbursts, optimizing carrier mobility through simulation is indispensable for meeting the evolving demands of modern electronics. Thus, it is vital to calculate the fundamental parameters of semiconductor compounds, such as band-structure, phonon dispersion relation, and many more, before fabricating a prototype device to theoretically optimize the material and thus improve the entire development process.
We use density functional Theory (DFT) to calculate band structure and density functional perturbation theory (DFPT) to calculate the phonon properties of a compound. A typical challenge of DFT simulation in semiconductor compounds lies in accurately calculating the band structure of the system, which can ensure reliable charge transport properties. This is not feasible using typical exchange-correlation functionals such as LDA and GGA since they often underestimate the electronic band gap. At the same time, several approaches exist to address this issue and improve band structure description. However, these methods are often constrained by high computational costs in complex systems such as alloys and the lack of implementation and support in DFPT calculations. One potential approach is the Hubbard correction via the DFT+U method. Within this approach, the Hubbard correction acts selectively on certain manifolds through projectors on the corresponding states. In contrast, electrons in other states are treated using standard exchange-correlation functionals such as LDA or GGA. In our calculations, we exploit a Bayesian optimization scheme applied to the band structures to find ideal U parameters. This method can improve band structure (see Fig. 1) and phonon properties through DFPT+U calculations (see Fig. 2) with a reasonable computational cost.
To compute phonon-limited carrier mobility, the linearized Boltzmann transport equation is solved using an iterative approach (ITA-BTE). In this approach, the essential quantity is the electron-phonon coupling matrix with its elements depending on the momentum quantum numbers k and q. The elements of this matrix can be obtained accurately using DFPT simulations. However, obtaining credible charge transport results in this approach relies on calculating these elements on an exceedingly dense grid of k and q points, which is computationally impractical. An efficient solution involves utilizing maximally localized Wannier functions, enabling interpolating these elements for a dense k and q grid using values obtained for coarse grids via DFT and DFPT. (see Fig. 3). This approach yields reliable results with an efficient computational cost.
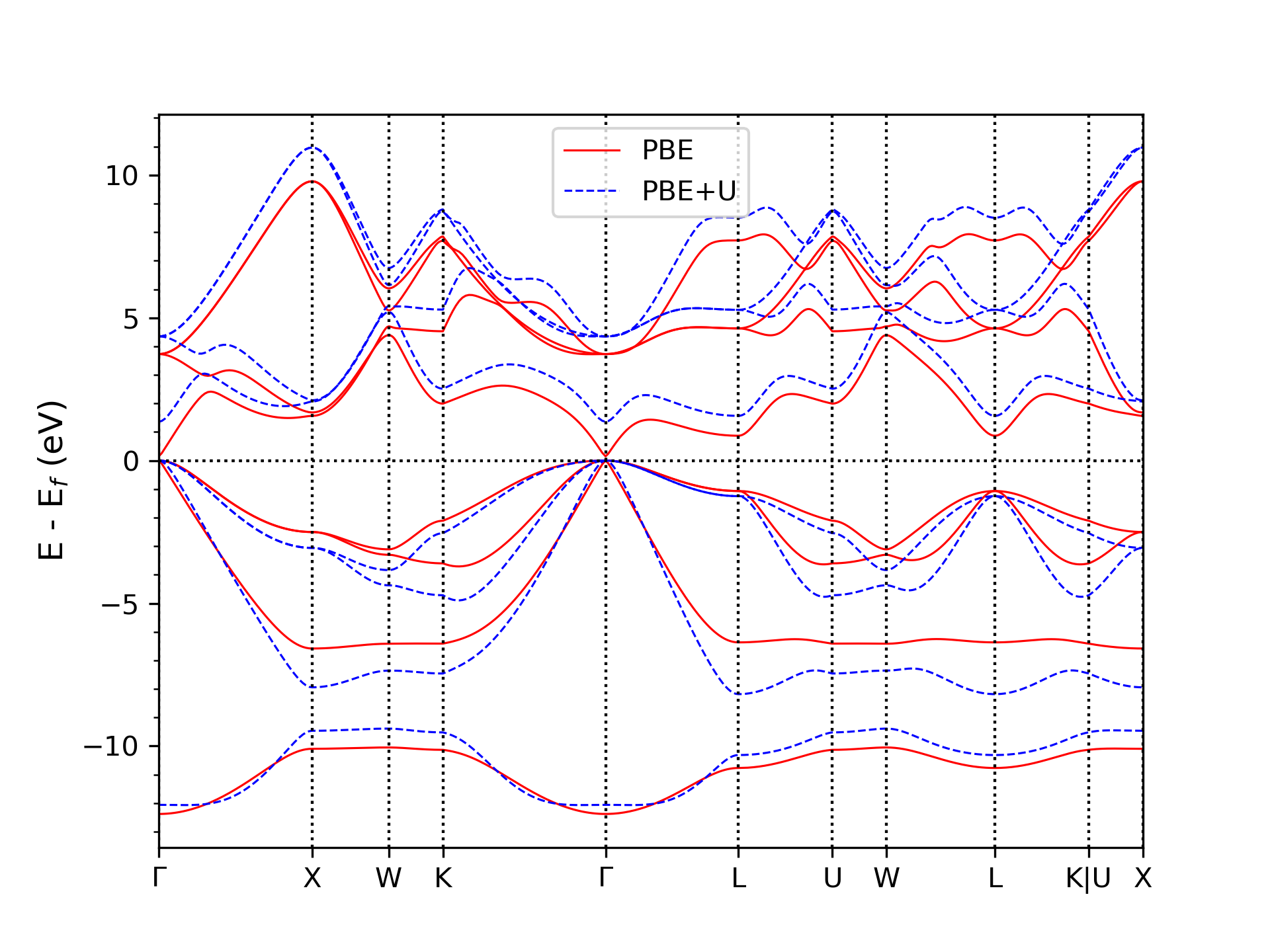
Fig. 1: Band structure of GaAs using PBE and PBE+U methods.
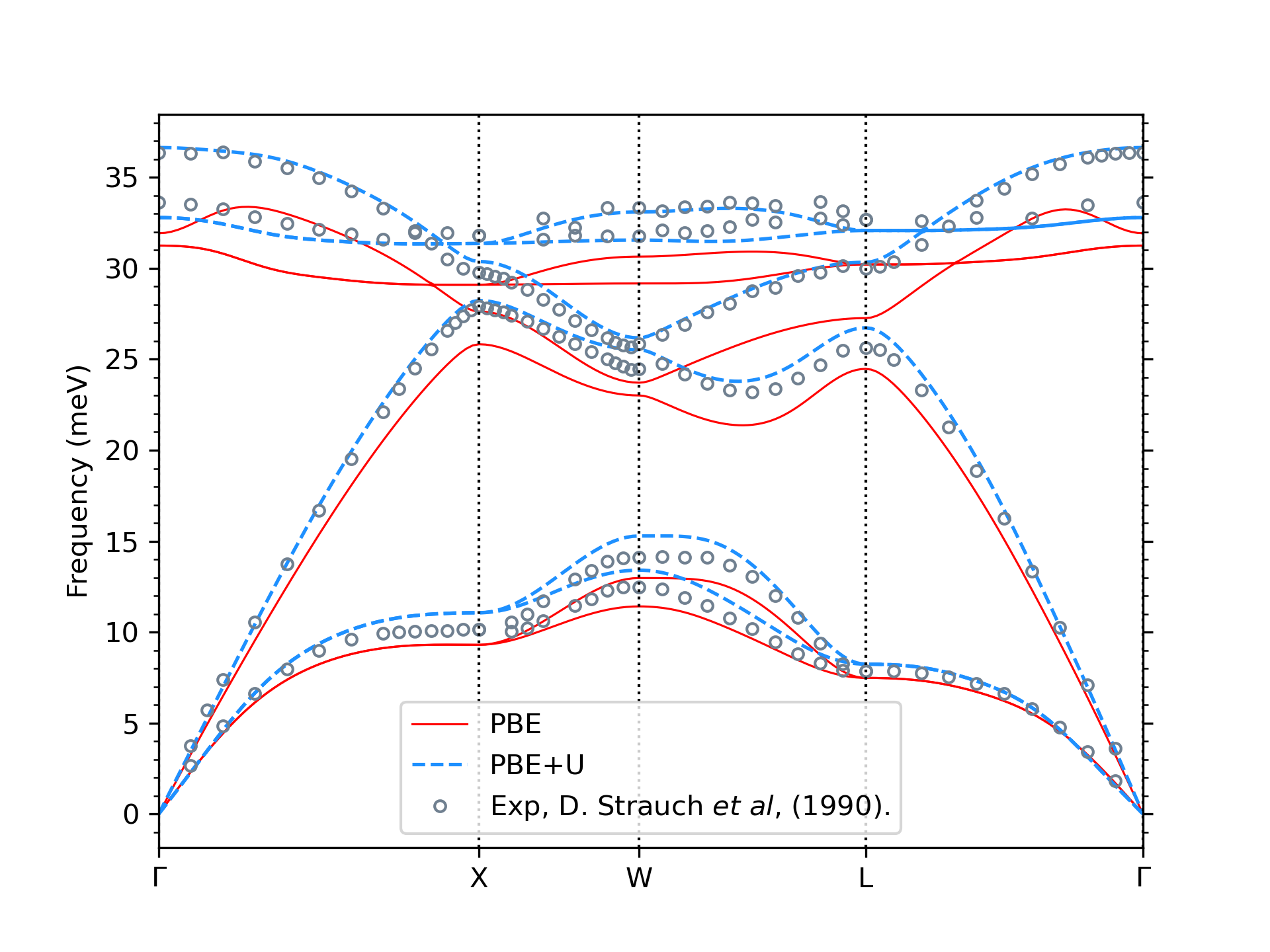
Fig. 2: Phonon Dispersion of GaAs using PBE and PBE+U methods compared to the experimental data.
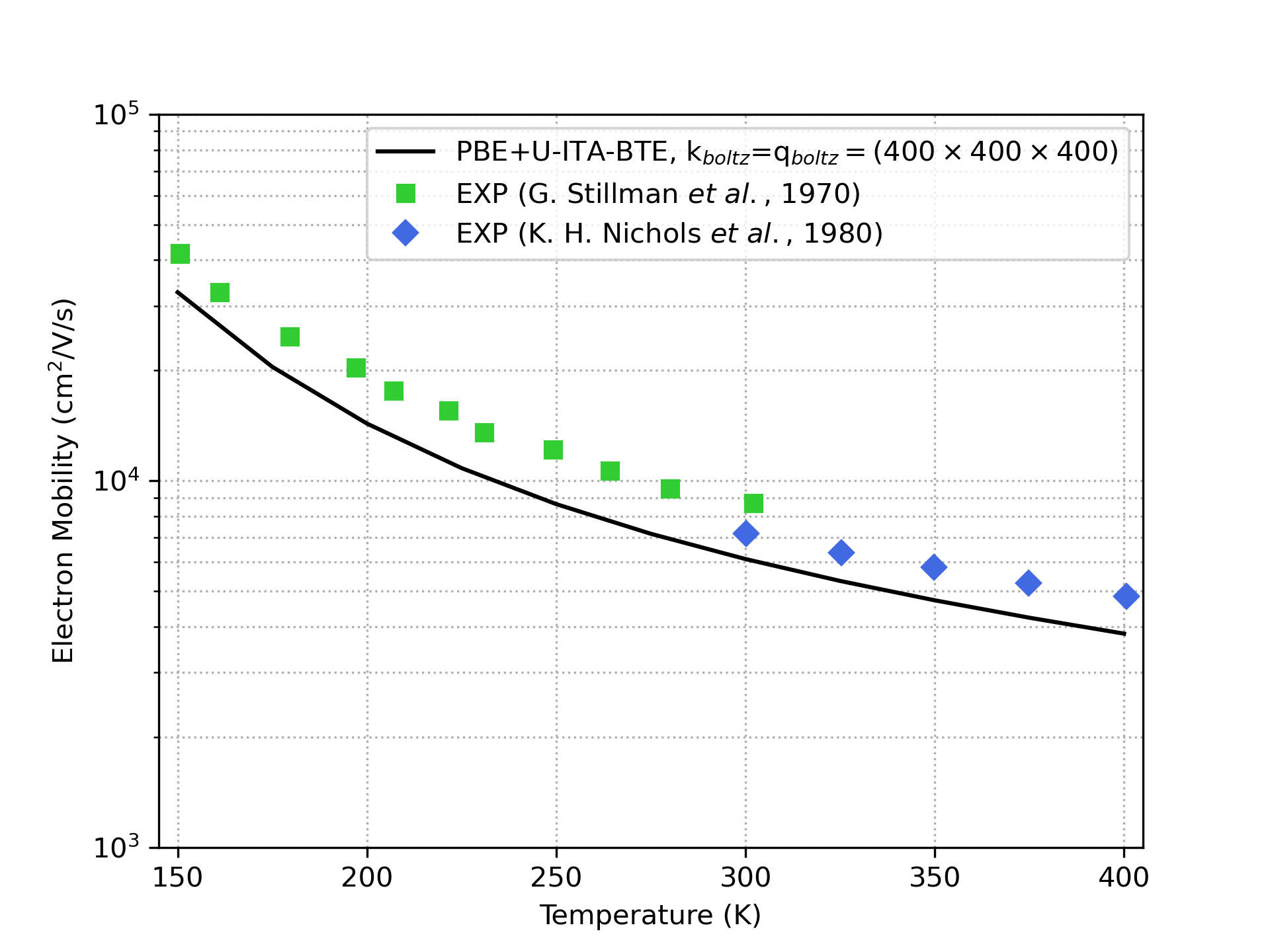
Fig. 3: Electron mobility of GaAs computed as a function of temperature using PBE+U and ITA-BTE compared with experimental data. The electron-phonon coupling matrix elements were calculated using maximally localized Wannier functions interpolation from a coarse k grid of (16×16×16) and q grid of (8×8×8) to a (400×400×400) grid for both k and q.