
 |
|
||||
BiographyMohammad Dehghani is from Isfahan, Iran, where he has finished his bachelor's and Master's degrees in Solid-state Physics. In late 2013, he moved to Leoben, Austria and started working as a research assistant at Materials Center Leoben (MCL) Forschung GmbH. After receiving his PhD in computational solid-state physics from the University of Graz in late 2017, he continued working at MCL as a Postdoc for two more years. His scientific and research background focuses on the Density Functional Theory (DFT) simulations of inter-metallic alloys. He has years of experience working on projects focused on computational code development and application on various alloys. He has joined the Institute for Microelectronics in January 2023 and is working on ab initio simulation of quaternary semiconductor alloys. |
|||||
Ab initio Calculation of Phonon-limited Electron Mobility in Indium Arsenide
We present a first-principles study of phonon-limited electron mobility in Indium Arsenide (InAs), a narrow-gap polar semiconductor with high electron mobility and strong technological relevance. Our computational framework combines density functional theory (DFT), density functional perturbation theory (DFPT), and Wannier interpolation of electron-phonon matrix elements to solve the linearized Boltzmann transport equation (LBTE) iteratively, as is implemented in Perturbo code. This approach enables an accurate and fully ab initio treatment of electron-phonon scattering processes.
It is well known that standard DFT calculations using LDA or GGA exchange-correlation functionals systematically underestimate the band gap and misrepresent the effective mass in semiconductors. These deficiencies affect not only electronic properties but also phonon dispersions—particularly in polar materials, where electron-phonon coupling and scattering rates are sensitive to the band structure and longitudinal optical phonon frequency at the Γ point. These limitations are especially critical for InAs, which has a narrow band gap and an extremely small conduction band effective mass, making accurate mobility predictions highly sensitive to this latter property.
To address these limitations, we employ the DFT+U method, where the Hubbard U parameter is optimized using a Bayesian approach. By targeting both the band structure and phonon frequencies at the Γ point, we identify a set of U values that yields an improved band gap, effective mass, and phonon frequencies at the conduction band minimum that aligns with experimental values. Using these refined electronic and vibrational properties, we compute the phonon-limited electron mobility by solving the LBTE, obtaining results in excellent agreement with experimental measurements.
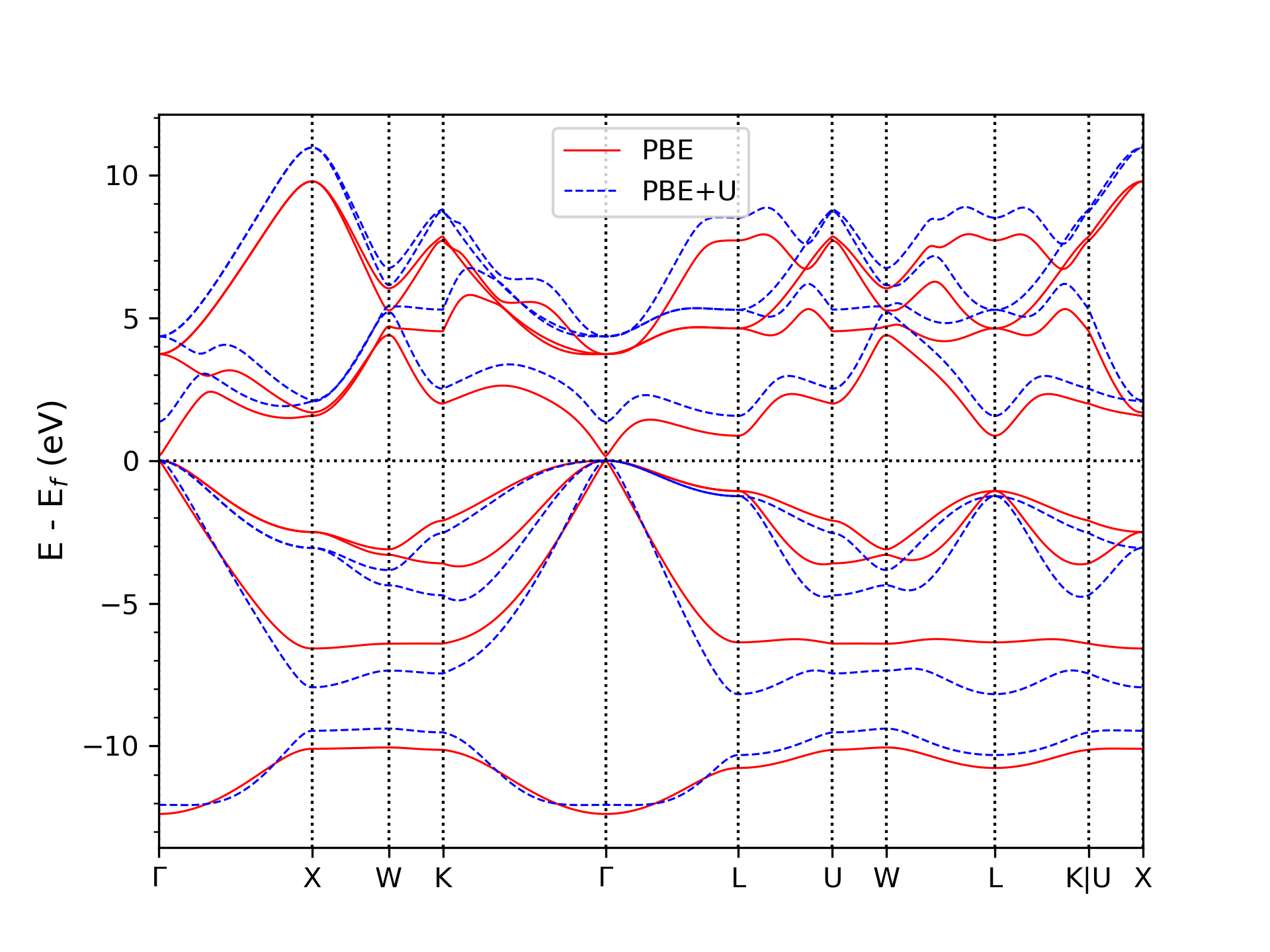
Fig. 1: Temperature-dependent electron mobility of InAs computed by solving the LBTE iteratively based on band structure and phonon properties obtained from PBE+U method, compared with experimental measurements. Electron-phonon coupling matrix elements were interpolated using maximally localized Wannier functions from coarse k- and q-point grids (16×16×16 and 8×8×8, respectively) to dense grids of (400×400×400) for both k and q.