3.8.1 The Empirical Pseudopotential Method
The pseudopotential theory is based on an ansatz which separates the total wave
function into an oscillatory part and a smooth part, the so called pseudo wave
function. The strong true potential of the ions is replaced by a weaker
potential valid for the valence electrons, the pseudopotential
 ,
which approaches the unscreened Coulomb potential of the Si
,
which approaches the unscreened Coulomb potential of the Si ion at
large values of
ion at
large values of  (see Figure 3.14). This replacement can be justified
mathematically and shown to reproduce correctly the conduction and valence band
states [Cohen89].
(see Figure 3.14). This replacement can be justified
mathematically and shown to reproduce correctly the conduction and valence band
states [Cohen89].
Figure 3.14:
Schematic plot of a pseudopotential in real space (left) and reciprocal space (right).
|
|
The one-electron Schrödinger equation is replaced by a pseudo-wave equation
 |
(3.101) |
with
 denoting the pseudo wave function and
the pseudopotential. This equation can be used to calculate
physical properties of semiconductors which are dependent on the valence and
conduction electrons only. Since pseudopotentials are only small
perturbations, the energy bands are expected to be similar to those of nearly
free electrons and an ansatz where the pseudo wave function is expanded into a
sum of plane waves can be used
denoting the pseudo wave function and
the pseudopotential. This equation can be used to calculate
physical properties of semiconductors which are dependent on the valence and
conduction electrons only. Since pseudopotentials are only small
perturbations, the energy bands are expected to be similar to those of nearly
free electrons and an ansatz where the pseudo wave function is expanded into a
sum of plane waves can be used
 |
(3.102) |
to diagonalize (3.101). Here,
 is a general reciprocal
lattice vector as given in (3.24) and Dirac's notation
is a general reciprocal
lattice vector as given in (3.24) and Dirac's notation
 is applied to denote the plane wave with wave
vector
is applied to denote the plane wave with wave
vector
 . The coefficients
. The coefficients
 and the eigenvalues
and the eigenvalues
 can be determined from the solution of the secular equation
can be determined from the solution of the secular equation
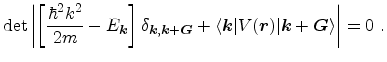 |
(3.103) |
The matrix elements of the pseudopotential are given by
 |
(3.104) |
Here,
 is a lattice vector in the real lattice,
is a lattice vector in the real lattice,  is the number of
atoms in the primitive unit cell3.2and
is the number of
atoms in the primitive unit cell3.2and  denotes its volume. The matrix elements are determined by the Fourier
components
denotes its volume. The matrix elements are determined by the Fourier
components
 of the pseudopotential
of the pseudopotential
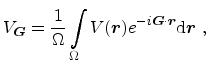 |
(3.105) |
which are frequently called form factors of the pseudopotential. If there is
more than one atom in the primitive unit cell, a structure factor is introduced
which depends on the relative position
 of the respective atom in
the primitive unit cell. The structure factor
of the respective atom in
the primitive unit cell. The structure factor
 is defined as
is defined as
 |
(3.106) |
where N denotes the number of atoms in the primitive unit cell. The
pseudopotential
can be expressed in terms of the structure factor
and the form factors by
 |
(3.107) |
In crystals with a diamond structure there are two atoms at the positions
 and
and
 in the primitive unit cell. By taking the
midpoint between the two atoms in the unit cell as origin, the positions of the
atoms are given by
in the primitive unit cell. By taking the
midpoint between the two atoms in the unit cell as origin, the positions of the
atoms are given by
 and
and
 . Thus, the structure factor is given by
. Thus, the structure factor is given by
 |
(3.108) |
In unstrained diamond structures the reciprocal lattice vectors in order
of increasing magnitude are (in units of
 ):
):
Form factors with reciprocal lattice vectors larger than
 are neglected, since typically
decreases as
are neglected, since typically
decreases as  for large
(see Figure 3.14). Assuming that the
atomic pseudopotentials are spherically symmetric
for large
(see Figure 3.14). Assuming that the
atomic pseudopotentials are spherically symmetric
 , the form factors only depend on the absolute value of the
reciprocal lattice vector. The form factor belonging to
, the form factors only depend on the absolute value of the
reciprocal lattice vector. The form factor belonging to
 shifts the
entire energy scale by a constant value, and can therefore be set to zero. The
form factors belonging to the reciprocal lattice vectors
shifts the
entire energy scale by a constant value, and can therefore be set to zero. The
form factors belonging to the reciprocal lattice vectors
 have an
absolute value of
have an
absolute value of
 and are conventionally
labeled
and are conventionally
labeled  . Since the structure factor of the reciprocal lattice vectors
. Since the structure factor of the reciprocal lattice vectors
 with magnitude
with magnitude
 vanishes,
vanishes,
 |
(3.109) |
the respective form factor  does not enter the pseudopotential
(3.107). Thus, only three pseudopotentials form factors ,
does not enter the pseudopotential
(3.107). Thus, only three pseudopotentials form factors ,  and
and  are required to calculate the band structure.
are required to calculate the band structure.
In Table 3.5 the parameters employed in the empirical
pseudopotential calculations are listed. They consist of three local form
factors
 , two parameters (
, two parameters ( ,
,
 ) to model the nonlocal correction, and two parameters (
) to model the nonlocal correction, and two parameters ( ,
,  )
entering the spin-orbit interaction term. The parameters coincide with the
parameter set provided in [Rieger93] with the exception of
)
entering the spin-orbit interaction term. The parameters coincide with the
parameter set provided in [Rieger93] with the exception of
 Ry and
Ry and
 Å
Å , which
have been adjusted in order to yield the desired split-off energy of
44 meV in the unstrained Si band structure. In the expansion of the
pseudo wave function (3.102) plane waves with modulo
, which
have been adjusted in order to yield the desired split-off energy of
44 meV in the unstrained Si band structure. In the expansion of the
pseudo wave function (3.102) plane waves with modulo
 were included, which guarantees results
converged to approximately 1 meV.
were included, which guarantees results
converged to approximately 1 meV.
E. Ungersboeck: Advanced Modelling Aspects of Modern Strained CMOS Technology
![\includegraphics[scale=1.0, clip]{inkscape/VqTot.eps}](img614.png)