
 |
|
||||
BiographyDiego Milardovich was born in Rosario, Argentina in 1992. He received his Master's degree in Electronic Engineering at the National University of Rosario in 2019. In February 2020, he joined the Institute for Microelectronics at the TU Wien, where he is currently working on his doctoral degree focusing on atomistic modeling of insulator defects. His previous experiences include 1 year as a research assistant at the Neuroscience and Biomedical Engineering department of Aalto University (Helsinki, Finland), 2 years as an engineering officer at Nestlé (Firmat, Argentina) and 3 months as a research engineer at Techint (Buenos Aires, Argentina). |
|||||
Machine Learning-Based Molecular Dynamics Accelerated by Neuromorphic Hardware
Molecular dynamics (MD) is a technique which allows simulating the behavior and evolution of atomic systems over time. It is an essential atomic-scale research method which has found use in a wide range of fields, including physics, biology, and nanotechnology. However, researchers employing this technique face a long-standing dilemma: accuracy versus efficiency. On the one hand, the ab initio evaluation, such as density functional theory (DFT), of the potential energy surface (PES) offer high accuracy, but at high computational costs, which limit their use to only small atomic systems. On the other hand, empirical potentials, which are hand-crafted approximations of the PES, are computationally more efficient, but they are not accurate enough for several applications.
During the last decade, machine learning (ML) interatomic potentials have emerged as promising alternatives to ab initio methods and empirical potentials. They aim to combine the high accuracy of the former with the low computational cost of the latter. ML interatomic potentials work by training an ML model, most frequently a neural network, to function as a computationally affordable surrogate model of the PES. Although these interatomic potentials have been successful in providing highly accurate results, their performance is currently severely limited by the hardware used to implement them. Presently, ML-based MDs are universally deployed on general-purpose computers based on the von Neumann architecture. In this architecture, the central processing unit (CPU) is separated from the main memory and is connected to it by a slow buffer, leading to the so-called von Neumann bottleneck. Running ML-based MD is a demanding task which requires the processing of data from thousands of atoms over millions of time-steps. When performing these simulations on a von Neumann computer, about 90% of the computing time (and energy) is consumed for the repeated transfer of data between the CPU and memory.
As an alternative to von Neumann computers, brain-inspired, or neuromorphic, architectures stand out for their co-location of the CPU and main memory. This characteristic makes neuromorphic computing an ideal candidate as a hardware accelerator for ML-based MD. Using the open-access tools provided by Mo et al., we developed a ML interatomic potential for Ge, with the goal of studying the physical mechanisms behind Ge-based memristors. As shown in Fig. 1, the MD simulations run on a hybrid platform, where the PES is evaluated in a neuromorphic or non-von Neumann (NVN) accelerator, implemented in a Field Programmable Gate Array (FPGA), while the remaining tasks are performed on a standard computer. Our preliminary results show that, using an equal number of CPU cores, this approach is about 100 times faster than a Gaussian Approximation Potential (GAP) and about 100,000 times faster than DFT. Moreover, this approach is highly accurate, as can be seen in the results shown in Fig. 2, where an amorphous Ge model, created by running a melt-and-quench MD with this method, is compared to a DFT-relaxed reference. The radial distribution function (RDF) and structure factor obtained from neutron scattering are computed for both atomic structures and the results are remarkably similar.
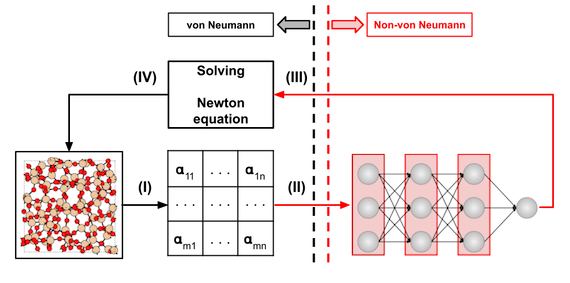
Fig. 1: Workflow to run the MD simulations on a hybrid platform. A descriptor of the atomic configuration is computed (I), which is used by the neural network to evaluate the PES (II); the energy and forces are used to solve the Newton equation (III) and the positions of the atoms in the system are updated (IV). The process is repeated for the next time-step. The PES is evaluated within a neuromorphic accelerator (II-III), while the remaining computation requirements are fulfilled by a standard computer (I-IV).
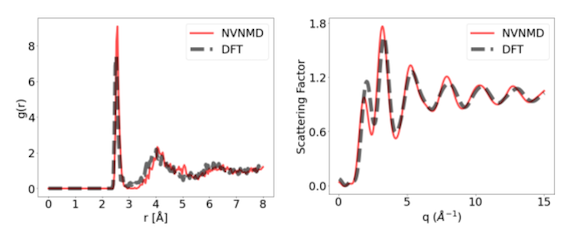
Fig. 2: Results of running a melt-and-quench MD for Ge with the proposed approach (NVNMD) and comparing the results to a DFT-relaxed reference. Left: RDF. Right: Structure factor obtained from neutron scattering.