Armchair graphene nanoribbons (AGNRs) have recently attracted much interest as they are recognized as promising building blocks for nanoelectronic devices [38]. AGNRs, a type of graphene nanoribbons (GNRs) with armchair edges, introduce a tunable band-gap which is suitable for electrical and optical applications [70].
Single-layer hexagonal boron nitride (h-BN) and boron nitride nanoribbons (BNNRs), which are regarded as the III-V analogs of graphene and GNRs, respectively, have been synthesized and studied in recent years [129, 130, 131, 132]. Theoretical and experimental results have shown that the sp2 bonding in the BN lattice gains an ionic character due to different electronegativity of B and N, that causes the optical and electronic properties of BN and BNNRs to be substantially different from that of graphene and GNRs [131]. Unlike graphene, which is a zero-gap material, h-BN has a wide bandgap of approximately 5.9eV and shows good insulating behavior [133].
BNNRs are expected to be produced using a single-layer h-BN as the starting material [131]. The similarity of the crystal structures of BN and graphene gives rise to thermodynamically stable two-dimensional structures containing isolated regions of graphene and BN [134, 132]. It has been shown that AGNRs embedded in BN sheets (AGNRs/BN) are semiconductors [132]. Due to the relatively large ionicities of boron and nitrogen, BN-confined AGNRs exhibit a generally larger bandgap compared to H-passivated AGNRs [135, 136]. These hybrid C-BN structures can be synthesized by approaches such as thermal catalytic CVD methods [134].
A relatively large bandgap of graphene nanoribbons incorporated in a BN lattice renders them as suitable candidates for opto-electronic applications. Structures composed of GNRs and BNNRs introduce more flexibility for electronic and opto-electronic applications. In this chapter, we investigate for the first time a theoretical study of the optical properties of graphene and graphene/BN nanoribbons.
Density functional theory (DFT) is a quantum-mechanical method used in chemistry and physics to calculate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases [137, 138]. With this theory, the properties of a many-electron system can be determined by using functionals, i.e. functions of another function, which in this case is the spatially dependent electron density. Hence the name density functional theory comes from the use of functionals of the electron density [139]. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry [140].
DFT has been very popular for calculations in solid-state physics since the 1970s [138]. However, DFT was not considered accurate enough for calculations in quantum chemistry until the 1990s, when the approximations used in the theory were greatly refined to better model the exchange and correlation interactions. In many cases the results of DFT calculations for solid-state systems agree quite satisfactorily with experimental data [141, 142]. Computational costs are relatively low when compared to traditional methods, such as Hartree-Fock theory and its descendants based on the complex many-electron wave function.
The major problem with DFT is that the exact functionals for exchange and correlation are not known except for the free electron gas. However, approximations exist which permit the calculation of certain physical quantities quite accurately [141]. An approximation to the exchange-correlation term is used. It is called the local density approximation (LDA). For any small region, the exchange-correlation energy is the approximated by that for jellium of the same electron density. In other words, the exchange-correlation hole that is modelled is not the exact one — it is replaced by the hole taken from an electron gas whose density is the same as the local density around the electron [143].
In physics, LDA is the most widely used approximation, where the functional depends only on the density at the coordinate where the functional is evaluated [144]
| EXCLDA[n]= | ∫ | єXC(n)n ( |
| ) d3r. (1) |
The local spin-density approximation (LSDA) is a straightforward generalization of the LDA to include electron spin:
| EXCLSDA[n↑,n↓]= | ∫ | єXC(n↑,n↓)n ( |
| )d3r. (2) |
Highly accurate formula for the exchange-correlation energy density єXC(n↑,n↓) have been constructed from quantum Monte Carlo simulations of Jellium [145]. Jellium, also known as the uniform electron gas or homogeneous electron gas, is a quantum mechanical model of interacting electrons in a solid where the positive charges (i.e. atomic nuclei) are assumed to be uniformly distributed in space whence the electron density is a uniform quantity as well in space [146].
The interesting point about this approximation is that although the exchange-correlation hole may not be represented well in terms of its shape, the overall effective charge is modelled exactly [146]. This means that the attractive potential which the electron feels at its centre is well described. Not only does the LDA approximation work for materials with slowly varying or homogeneous electron densities but in practise demonstrates surprisingly accurate results for a wide range of ionic, covalent and metallic materials [146].
An alternative, slightly more sophisticated approximation is the generalised gradient approximation (GGA) [147, 148, 149] which estimate the contribution of each volume element to the exchange-correlation based upon the magnitude and gradient of the electron density within that element. GGA are still local but also take into account the gradient of the density at the same coordinate:
| EXCGGA[n↑,n↓]= | ∫ | єXC(n↑,n↓, |
| n↑, |
| n↓) n ( |
| ) d3r. (3) |
Using GGA very good results for molecular geometries and ground-state energies have been achieved.
In this work, the imaginary part of the dielectric function is calculated by DFT to investigate the optical properties of studied structures [150]. If the incident light is assumed to be polarized along the transport direction (x-axis), the imaginary part of the dielectric function in the linear response regime is given by [150]
|
where ℏω is the energy of the incident photons and pc,v are the momentum matrix elements for optical transitions. The Dirac delta is approximated by a Gaussian function with a broadening factor of 0.1 eV.
The summation in Eq. 4 can be converted into an energy integration by introducing the joint density of states (JDOS) defined as
| Dj(ℏω) = |
| ∫ |
|
| , (5) |
where Skx is the constant energy surface defined by Ec(kx)−Ev(kx)=ℏω.
The real part of the dielectric function εr(ω) can be evaluated from the imaginary part using the Kramers-Kronig relation [150]. The interband dielectric function is related to the optical conductivity by ε(ω)=1+4π i σ(ω)/ω [151].
For DFT calculations, we employed the Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA) package [152] with the following parameters: double-ζ basis set with additional orbitals of polarization for total energies and electronic band-structures calculations, the generalized gradient approximation method, Perdew-Burke-Ernzerhof (PBE) as the exchange-correlation function, and the Troullier-Martins scheme for the norm-conserving pseudopotential calculations [135]. A grid cutoff of 210 Ry is used and the Brillouin zone sampling is performed by the Monkhost pack mesh of k-points. A mesh of (128×8×1) has been adopted for discretization of k-points and a broadening factor of 0.1eV is assumed for the joint density of states calculation, and the convergence criterion for the density matrix is taken as 10−4.
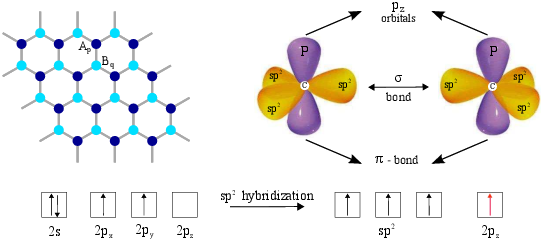
In graphene three σ bonds hybridize in an sp2 configuration, whereas the other 2pz orbital, which is perpendicular to the graphene layer, forms π covalent bonds. Each atom in an sp2-coordination has three nearest neighbors, located acc=1.42 Å away, see Fig. 3.1. It is well known that the electronic and optical properties of GNRs are mainly determined by the π electrons [153]. To model these π electrons, a nearest neighbor tight-binding approximation has been widely used [51, 154]. Based on this approximation the Hamiltonian can be written as:
| , (6) |
| Ap⟩ and | Bq⟩ are atomic wave functions of the 2pz orbitals centered at lattice sites and are labeled as Ap and Bq, respectively, ⟨ p,q ⟩ represent pairs of nearest neighbor sites p and q, t=⟨ Ap | H | Bq ⟩ ≈ −2.7eV is the transfer integral, and the on-site potential is assumed to be zero.
To study AGNR/BN, however, a TB model incorporating at least two nearest neighbors is required [135]. Reference [135] has shown that the band structure of AGNRs/BN can be calculated within the desired precision assuming the orthogonality of atomic orbitals and considering the effect of more nearest neighbors for each atom. Reference [155] shows that taking the effect of the first three neighboring atoms into consideration results in a good agreement with first principles calculations. Also, considering the second nearest neighbor carbon atoms in TB calculations will shift the dispersion relation by a constant value, [155] thereby affecting the optical transition rules. Therefore, up to second nearest neighbors are included in our work employing the parameters reported in Ref. [135].
To investigate the optical response of AGNRs/BN, the incident light is assumed to be polarized along the transport direction (x-axis). Particularly, it is shown that the photocurrent is maximized for photons polarized along the longitudinal direction of the structure, [156] which are the main source for interband transitions [151, 157, 158].
Over the past decade the non-equilibrium Green’s function formalism has been widely employed to investigate various nano electronic devices [159]. Four types of Green’s functions are defined as the non-equilibrium statistical ensemble averages of the single particle correlation operator. The greater Green’s function, G>, and the lesser Green’s function, G<, deal with the statistics of carriers. The retarded Green’s function, GR, and the advanced Green’s function, GA, describe the dynamics of carriers. Under steady-state condition the equation of motion for the Green’s functions can be written as [159]:
| GR(E) = | ⎡ ⎣ | ⎛ ⎝ | E + i0+ | ⎞ ⎠ | I − H − Σ R(E) | ⎤ ⎦ |
| (7) |
| G≷(E) = GR(E) Σ(E)≷ GA(E) (8) |
where H is the Hamiltonian matrix. In this formalism the effect of various interactions is included in the self-energy term:
| (9) |
where Σ1 and Σ2 are the self-energies of the left/right contacts and Σs is the scattering self-energy. The self-energy due to electron-photon interaction is considered in this work. The Hamiltonian of the electron-photon interaction can be written as [160, 161]:
| Ĥe-ph = |
|
| Â · ⟨ i | p | j ⟩ , (10) |
where p is the momentum operator and  is the vector potential. In second quantization the vector potential is given by
| Â = a |
|
| ⎛ ⎝ | be−iω t + b†e+iω t | ⎞ ⎠ | (11) |
The direction of  is determined by the polarization of the field, which is denoted by the unit vector a.
As atomic orbitals in tight-binding are unknown, to evaluate the matrix elements of the momentum, it is common to use the gradient approximation [162] where the following commutator relation is used p = (i m0/ℏ) [Ĥ,r].
Based on this approximation, for a transition from a state | m ⟩ to a final state | l ⟩, the momentum matrix elements (pxl,m=⟨ l | px | m ⟩) are
| pxl,m=(im0/ℏ)⟨ l| Ĥx−xĤ | m⟩. (12) |
The momentum matrix elements are therefore given by [163]
| pxl,m=(xm−xl) |
| ⟨ l| Ĥ | m⟩. (13) |
Therefore, one can rewrite the electron-photon interaction Hamiltonian as:
| Ĥe-ph = |
| Ml,m | ⎛ ⎝ | be−iω t + b†e+iω t | ⎞ ⎠ | âl† âm (14) |
| Ml,m = | ⎛ ⎝ | xm−xl | ⎞ ⎠ |
| ⎛ ⎜ ⎜ ⎝ |
| Iω | ⎞ ⎟ ⎟ ⎠ |
| ⟨ l|Ĥ0|m ⟩ (15) |
where xm denotes the position of the carbon atom at site m and Nω is the photon population number with the frequency ω.
The photon flux (Iω), is defined as the number of photons per unit time per unit area [160, 164]:
| Iω≡ |
| =Pop/(ℏω), (16) |
where єr and µr are the relative dielectric and magnetic susceptibility, V is the volume, and Pop is the incident power flux. The incident light is assumed to be monochromatic with a power of Pop=100kW/cm2, and polarized along the longitudinal axis, see Fig. 5.1.
We employed the lowest order self-energy of the electron-photon interaction based on the self-consistent Born approximation [165]:
| (17) |
where the first term corresponds to the excitation of an electron by the absorption of a photon and the second term corresponds to the emission of a photon by de-excitation of an electron.
The interband optical matrix element for a transition from an eigenstate in the valance subband to another eigenstate in the conduction subband is calculated in Sec. 3.1.3. These matrix elements determine the selection rules for optical transitions [166]. A zero matrix element means a forbidden transition. To determine a transition rule, it is sufficient to determine the symmetry of the transition matrix element. If the symmetry of this element spans the totally symmetric representation of the point group to which the unit cell belongs then its value is not zero and the transition is allowed. Otherwise, the transition is forbidden. Assuming a uniform potential profile across the ribbon’s width, the subband’s wave functions are either symmetric or anti-symmetric along the y-axis direction (ψc/v(−y)=±ψc/v(y)). Therefore, the momentum matrix elements are non-zero for interband-transitions from the symmetric (anti-symmetric) to the symmetric (anti-symmetric) wave functions. This transition rule results in transitions from subbands with odd (even) to odd (even) indices in AGNRs/BN, which is described later.
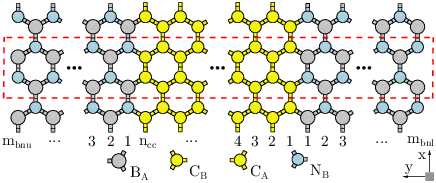
Figure. 3.2 shows the structure of an AGNR/BN represented by AGNRnccBNmbn, where ncc is the number of carbon dimers in the unit cell of the AGNR and mbn=mbnu+mbnl is the summation of the upper and lower BNNRs dimers.
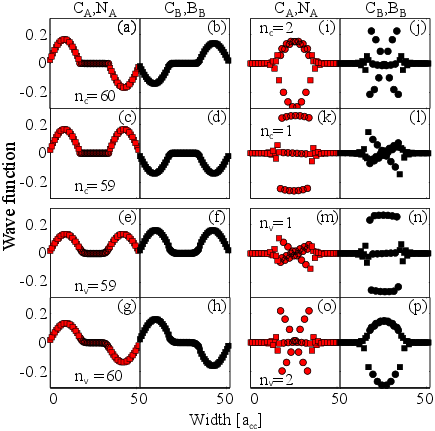
The wave functions at the sublattices A and B of an AGNR20BN40 at kx=0 are shown in Fig. 3.3. CA/B, NA/B, and BA/B represent the components of the wave functions at a carbon, nitrogen, and boron atom at some sublattice A or B. The wave function of each subband is the summation of the wave functions at these two sublattices. For example, the wave functions for the subband indices nv=60 (Fig. 3.3(g) and (h)), nc=2 (Fig. 3.3(i) and (j)), and nc=60 (Fig. 3.3(a) and (b)) are anti-symmetric. Therefore, as discussed before, the matrix elements are non-zero for transitions from nv=60 to nc=2 and nc=60. The transitions from nv=59 to nc=1 and nc=59 are also possible as their respective wave functions are symmetric. With the same analysis, a transition from the highest valence subband to the lowest conduction subband (nv=1 to nc=1) is possible in AGNRs/BN, see Fig. 3.3(m) and (n) and Fig. 3.3(k) and (l).
To obtain the transition rules, momentum matrix elements should be calculated. The total wave functions of the system is given by [167]
| | ψ ⟩ = CA | ψA ⟩ + CB | ψB ⟩ . (18) |
The Bloch wavefunctions | ψA⟩ and | ψB⟩ can be expressed as a linear combination of atomic wavefunctions of 2pz orbitals | Ap⟩ and | Bq⟩. Due to translational invariance along the x direction one obtains
| | ψA⟩ = |
|
| ei kx xpA φp| Ap⟩ , | ψB⟩ = |
|
| ei kx xpB φp| Bp⟩ , (19) |
where ΩA/B are the normalization factors, N is the number of A and B sublattices in the unit-cell of the GNR, xpA/B are the x-positions of the pth A/B-type carbon atom, φp is the y direction component of the wavefunctions at the pth lattice site. We impose hard-wall boundary conditions [168] at the edges, φ0=0 and φN+1=0. Therefore, one can assume that the component of the wavefunctions in the y direction form standing waves
| φp=sin |
| =sin |
| , n=1,2,…,N. (20) |
where n is the band index. For convenience the notation θ=π/(N+1) is introduced. Assuming the normalization condition ⟨ψA| ψA⟩=⟨ψB| ψB⟩=1,[168] the prefactors are obtained as ΩA=ΩB=Nx(N+1)/2, where Nx is the number of unit cells along the x direction. For a perfect and uniform ribbon we just need to perform the calculations over one unit-cell, therefore, from here on we assume Nx=1. Finally, the coefficients CA and CB in Eq. 18 are found by solving the Schrödinger equation, H|ψ⟩=E|ψ⟩, resulting in CB=± CA e−iϕn(kx), where ϕn(kx) is defined as
| eiϕn(kx)= |
| , (21) |
in which fn(kx)=eikxacc+2e−ikxacc/2cos(nθ). To satisfy the normalization condition, | CA|2+| CB|2=1, one can choose CA=1/√2 and CB=± e−ϕn(kx)/√2. The wavefunction is given by
| | ±, n, kx⟩ = |
| ⎡ ⎢ ⎢ ⎣ |
| ⎛ ⎝ | eikxxpAsin |
| | Ap⟩ ∓ e−iϕn(kx)eikxxpBsin |
| | Bp⟩ | ⎞ ⎠ | ⎤ ⎥ ⎥ ⎦ | , (22) |
where the notation | ±, n, kx⟩ ≡ | ψn±(kx)⟩ is introduced and ± denote the conduction and the valence bands, respectively.
Based on Eq. 22, one can approximate the wave functions of an H-AGNR at pth atomic site with sin(nθ p) functions, where n is the subband index and θ = π / (N+1). Considering such wave functions, the momentum matrix elements are
| pn,m(kx) = |
|
| tacc | ⎡ ⎢ ⎢ ⎣ |
| sin |
| sin |
| ⎤ ⎥ ⎥ ⎦ | Fn,m(kx). (23) |
According to this equation, only transitions between valence and conduction subbands with the same band-index are allowed (shown in Appendix A). As shown in Fig. 3.3, the wave functions for AGNRs/BN are not necessarily a single sinusoidal function. The Fourier series of these wave functions contain sinusoidal functions with different arguments and coefficients, which results in more allowed interband-transitions compared to H-AGNRs. Non-zero terms and the symmetry properties of the AGNR/BN wave functions indicate that the interband-transitions between subbands with the same parity are allowed (odd to odd and even to even). This transition rule is more restricted for conventional H-AGNRs where the wave functions consist of complete sine terms (see Eq. 5) [169]. The rules are also different from that of ZGNRs where interband-transitions from subbands with odd (even) indices to subbands with even (odd) indices are allowed. [170].
We compare our TB results with first principles calculations introduced in Sec. 3.1.1. The optical polarization vector is assumed to be along the transport direction similar to the assumption made in the TB calculation.
Figure 3.4(a) exhibits εi(ω) and the JDOS of AGNR20BN40.
In the energy range 0<ℏω<2 eV the JDOS has maxima at ℏω=0.3019, 0.62214, 0.92657, 0.91883, 1.2289,
1.5256, and 1.7349 eV. However, only four of these maxima (ℏω=0.3019, 0.91883,
0.92657, and 1.5256 eV) appear in εi(ω). From the electronic band-structure in Fig. 3.4(b) it can be shown that the peaks in εi(ω) are related to transitions from nv=1 to nc=1 (A), nv=1 to nc=3 (B), nv=2 to nc=2 (C), and nv=2 to nc=4 (D).
Disappeared peaks in εi(ω) are due to zero momentum matrix elements in Eq. 4. This transition rule confirms previous results which are explained by the symmetry properties of the wave functions.
Figure 3.4(a) compares the dielectric functions of an AGNR20BN40 obtained from TB and first principle calculations. Excellent agreement between these results confirms the transition rules obtained from TB calculations. The energy of the first peak matches well, however, the discrepancies increase for higher peaks. This behavior is related to the differences between the predicted energy-gaps obtained from SIESTA and TB at higher energies, see Fig. 3.4(b).
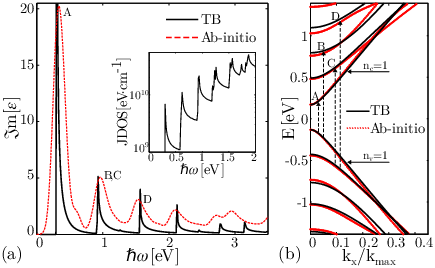
Figure 3.4: (a) The dielectric function of an AGNR20BN40 based on TB (solid line) and first principle calculations (dashed line). The inset shows the related JDOS using the TB model. (b) The electronic band-structure of an AGNR20BN40 from TB (solid line) and first principle calculations (red dotted line).
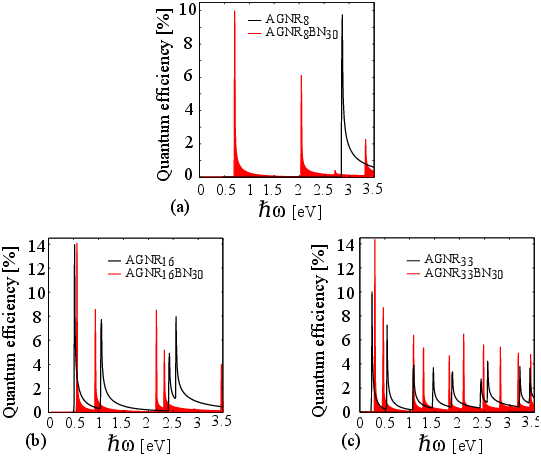
In order to investigate AGNRs/BN for photodetection application, we study the quantum efficiency defined as α=( Iph / q ) / (Pop / ℏ ω ), where Iph is the photo current and Pop is the incident optical power. We assumed that all absorbed photons contribute to the photo current, such that the quantum efficiency can be calculated from the dielectric function (Eq. 4). A quantum efficiency of 6−16 % for graphene is reported in Ref. [82] and a maximum quantum efficiency ranging from 9% to 11% is reported for H-AGNRs in Ref. [171]. Figure 3.5 shows the calculated quantum efficiency as a function of the incident photon energy at various GNR widths. The efficiency is maximized when the photon energy matches the bandgap of the nanoribbon (the first peak for each structure). Our results indicate a peak of quantum efficiency in the range of 14−15 % for AGNRs/BN. The quantum efficiencies of photodetectors based on AGNRs/BN and H-AGNRs are compared in Fig. 3.5. Due to the presence of more allowed transitions a wider absorption spectrum is achieved in AGNRs/BN compared to H-AGNRs. As a H-AGNR with index 8 is metallic, the first peak is related to the second energy-gap and appears at 2.88 eV whereas the AGNR8BN30 shows three peaks below that energy due to energy-gap opening, see Fig. 3.5(a). In Fig. 3.5, the quantum efficiency decreases for the first energies, but increases at higher energies, see for example the sixth peak for AGNR33BN30 (Fig. 3.5(c)). This is due to different effective masses of different subbands which affect the JDOS. According to Eq. 4, a larger JDOS leads to a larger absorption of photons and a higher quantum efficiency.
We also investigate the photoresponsivity given by ( Iph / Pop ). Our calculations give an upper limit for the photoresponsivities of 0.336 A/W, 0.239 A/W, and 0.202 A/W for photon energies near the bandgaps of AGNR8BN30, AGNR16BN30, and AGNR33BN30 respectively. Due to the higher quantum efficiency of AGNRs/BN compared to AGNRs, a higher photoresponsivity is obtained for the same input optical power of 107 W/m2.
In summary, we theoretically studied the optical properties of AGNRs/BN, employing TB calculations. We demonstrate that in AGNRs/BN only optical transitions from subbands with odd (even) indices to subbands with odd (even) indices are allowed. This transition rule is more restricted for AGNRs and completely different from that of ZGNRs. Our TB results are in agreement with first principle calculations which verifies the accuracy of our model. The applicability of AGNRs/BN as photodetectors is investigated. Our results indicate that due to more allowed transitions compared to conventional GNRs a larger photo current in AGNR/BN structures can be achieved. The results render AGNRs/BN as suitable candidates for infrared photodetectors and future optoelectronic applications.